


トランスレーショナル・ リサーチ部門
部門の紹介
当科では、群馬大学人を対象とする医学系研究倫理審査委員会の承認を得て、遺伝性循環器疾患の中で主に遺伝性不整脈症候群・心筋症(遺伝性QT延長症候群、ブルガダ症候群、カテコラミン誘発性多形性心室頻拍、不整脈源性右室心筋症など)の遺伝子解析を施行しております。
我々の研究の目的は、①疾患の原因となる遺伝子変異・多型を同定し、②遺伝子変異・多型により疾患を発症するメカニズム、疾患の分子病態を解明し、さらに、③得られた知見を実地診療に生かすことです。我々の研究は小規模ではありますが、地域に根ざしており、患者様から得られた遺伝情報を実地診療において患者様に最大限に還元する医療を実践しております。我々はこれまでに、遺伝性QT延長症候群、ブルガダ症候群、カテコラミン誘発性多形性心室頻拍、不整脈源性右室心筋症等において、新規変異を含む多くの変異を同定し、臨床像を解析し、さらに変異遺伝子の機能異常を解明することにより得られた多くの情報を発信してきました。我々の研究成果は、疾患の分子病態を解明し、致死性不整脈発症の予知・リスク層別化を可能とし、突然死予防につながるものと確信しております。
遺伝性循環器疾患と診断される患者様の数は年々増加しており、循環器疾患の診療において遺伝子診断/診療は、今後ますます重要になると考えられます。同時に、遺伝子診断/診療は「遺伝性」であるが故、倫理面を含め様々な問題を多く有しており、患者様の精神的ケアもとても重要です。我々は、患者様にエビデンスに基づく最先端の医療を提供していくとともに、医師、パラメディカル、遺伝子診療部が連携し、遺伝性疾患診療がかかえる課題を少しでも克服できる診療システムの構築を模索しております。
所属メンバー
不整脈診療に従事:
金古善明客員教授、田村峻太郎助教、長谷川寛助教
トランスレーショナルリサーチ(遺伝子解析、機能解析)に従事:
中島忠客員准教授、松井美紀さん(研究員)
遺伝子解析に関するお問い合わせ窓口
遺伝性不整脈症候群の遺伝子解析をご希望の方は、下記へご一報下さい。
群馬大学医学部附属病院 循環器内科
中島忠
e-mail: tnakajim@gunma-u.ac.jp
研究プロジェクトの紹介
次世代シークエンスの導入により、遺伝性QT延長症候群(LQTS)、ブルガダ症候群、カテコラミン誘発性多形性心室頻拍などの遺伝性不整脈症候群と関連する多くの遺伝子変異が同定され、また、疾患の病態も解明されつつあり、さらに、最適な治療法も確立しつつあります。特にLQTSでは遺伝子型により最適な治療法は異なることが明らかになっております。一方、特異及び多彩な表現型を呈する遺伝性不整脈症候群症例・家系も少なからず存在しますが、疾患と関連する遺伝子変異や病態は未解明のことが多く、治療法も確立していないのが現状です。
我々は、これまでに特異及び多彩な表現型を呈する遺伝性不整脈症候群症例・家系を多く経験し、次世代シークエンス(パネル解析、全エクソン解析)を駆使し、これらの症例・家系において多くの遺伝子変異を同定し得ております。さらに同定された遺伝子変異の機能異常を解明することにより遺伝性不整脈症候群の分子病態を解明し、将来の新規治療法を開発、確立するための知見を得ることを目的に研究を行っております。
最近の主な研究成果
特異及び多彩な表現型を呈したブルガダ症候群で同定されたSCN5A変異の機能異常の解明: fast inactivation 及びclosed-state inactivationの検討
ブルガダ症候群の心イベントの発症は安静時や睡眠時に多いことが知られております。しかしながら、我々は、洞機能不全、上室性頻脈性不整脈を合併し、運動中に心イベントを繰り返し発症した親子例を見出し(亜型ブルガダ症候群)、新規SCN5A R1632C変異(domain Ⅳ-segment 4: DⅣ-S4の変異)を同定し得ました(図1)。(Nakajima et al, Heart Rhythm 2015) パッチクランプ法による機能解析では、R1632C変異はナトリウム電流(INa)のfast inactivationからの回復が著明に遅延し、fast inactivationの安定性が増強していました。そのため、高頻度刺激によりR1632C-INaは著明な減衰を認めました。これらのことから、本症例では運動による心拍数上昇にともないINaがさらに減少し心イベントを発症したと考えられました。通常、ブルガダ症候群では、β刺激薬が抗不整脈作用を発揮し、β遮断薬は避けるべき薬剤とされておりますが、亜型ブルガダ症候群ではβ刺激薬は病態を悪化させる可能性や、逆にβ遮断薬の有効性が示唆されました。
さらに、最近、ブルガダ症候群、洞不全症候群、上室性頻脈性不整脈を呈した他の一症例に新規SCN5A N1541D変異(domain Ⅳ-segment 1: DⅣ-S1の変異)を同定し得ました(図1)。(Dharmawan et al, J Mol Cell Cardiol 2019) R1632C変異キャリアとN1541D変異キャリアは類似した表現型を呈したことから、その機能異常をパッチクランプ法にて解析し比較したところ、N1541Dは野生型と比べ、INaのsteady-state inactivation curve (SSIC)の著明な過分極シフトを認め、それは、closed-state inactivation:CSIの増強に起因していました(図2)。N1541Dと同様にSSICの著明な過分極シフトをきたしたR1632C変異(DⅣ-S4の変異)においてもCSIが増強し、CSIの速度は野生型と比べ軽度促進していましたが(図2)、CSIからの回復の速度は野生型と比べ著明に遅延していました(tau-野生型: 1.90±0.16 ms, R1632C: 53.0±2.5 ms, P<0.01 vs 野生型)(図3)。一方、N1541DのCSIの速度は野生型と比べ著明に促進していましたが(tau-野生型: 65.8±7.4 ms, N1541D: 13.7±1.1 ms, P<0.01 vs 野生型)、CSIからの回復の速度は野生型とほぼ同等でした(図3)。N1541D、R1632CはともにCSIの増強により著明なINa減弱をきたしましたが、そのメカニズムは異なることが明らかとなりました。
Nav1.4(骨格筋タイプ)では、N1366(Nav1.5ではN1541に相当)とR1457(Nav1.5ではR1632に相当)は近接しており、 チャネル活性化-不活性化プロセスで連結していると考えられていることから、Nav1.5のN1541とR1632でも同様と考えられます。両変異が異なるメカニズムによりCSIの増強をきたしたことは、Nav1.5の構造-機能連関に新たな知見をもたらしました。また、CSI増強によるINa減弱により多彩な表現型を呈することが説明可能であり、遺伝子型-表現型の解明の一助となると考えられました。
これらの研究成果は、臨床面では、遺伝性不整脈症候群における新規治療指針の基礎を構築し、また、基礎研究面では、イオンチャネルの構造-機能連関に新たな知見をもたらすなど、多方面に派生する可能性があります。
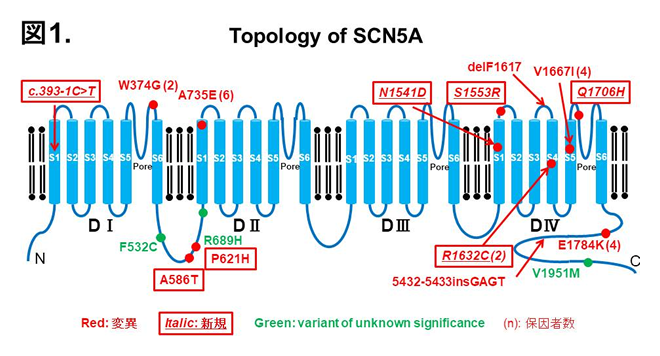
当院で同定した遺伝性不整脈症候群患者のSCN5A変異。
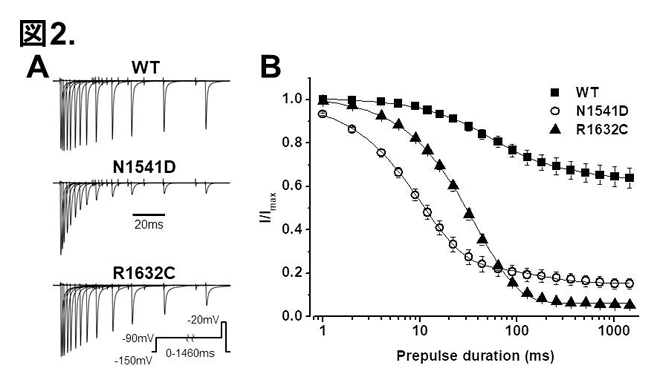
SCN5A野生型(WT), N1541D変異, R1632C変異のclosed-state inactivationの比較。
A: 挿入プロトコールでの電流記録
B: 脱分極パルス-90 mVでのclosed-state inactivation
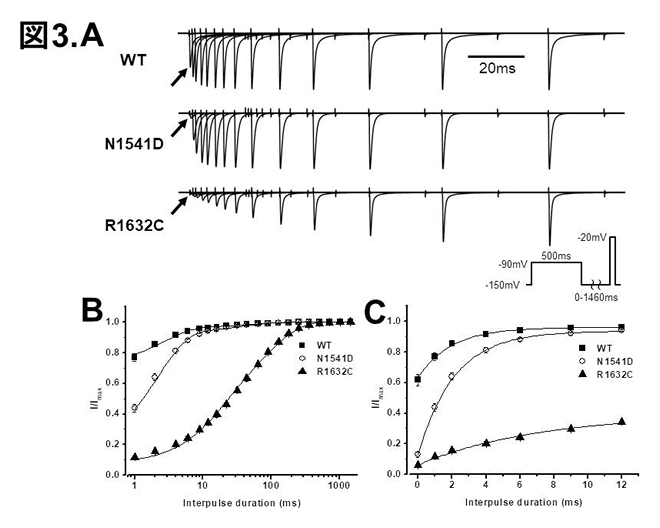
SCN5A野生型(WT), N1541D変異, R1632C変異のclosed-state inactivationからの回復の比較。
A: 挿入プロトコールでの電流記録
B: 過分極パルス(1-1460ms)でのclosed-state inactivationからの回復
C: 過分極パルス(0-12ms)でのclosed-state inactivationからの回復。
エピネフリン負荷で著明なQT延長をきたしたLQT3家系の同定及びSCN5A変異の機能異常の解明
我々は、エピネフリン負荷で著明なQT延長をきたしたLQTS家系(発端者は心肺停止蘇生後)にSCN5A V1667I変異を同定しました。(Nakajima et al, J Cardiovasc Electrophysiol in press) 通常、SCN5A変異によるLQTS (LQT3)は、運動やエピネフリン負荷では著明なQT延長はきたさないことから、本家系は遺伝子型-表現型の観点から特異であると考えられます。また、LQT3ではβ遮断薬の有効性はcontroversialですが、本変異のキャリアでは有効であることが示唆されます。野生型SCN5A、V1667I SCN5AをtsA201細胞に発現させ、パッチクランプ法にて機能解析を行った結果、V1667Iはナトリウム電流(INa)密度の増加、不活性化曲線の脱分極シフトによるwindow currentsの増加を認めました。さらに、protein kinase A activatorである8-CPT-cAMPを投与したところ、V1667Iは野生型に比し、window currentsの増加を認めました。SCN5A V1667I変異キャリアではエピネフリン負荷でQT延長をきたす分子メカニズムが解明され、また、本変異キャリアではβ遮断が有効であることが示唆されました。
新規心脳チャネル病の同定
我々は、発作性心房細動、早期再分極/J波(心臓チャネル病)と小児期にてんかん(脳チャネル病)を合併した10代姉妹(姉:19歳、妹:16歳時に突然死)を経験しました。発端者(姉)の全エクソン解析の結果、KCND3 V392I変異を同定し、妹も本変異を有していました。発端者にキニジンを投与したところ、心房細動発作の頻度は減少し、また、心電図上J波は減高しました。
本変異キャリアの臨床像の報告は乏しく、原因不明の突然死症例一例(20歳、男性)と、てんかん症例一例(5歳、男児)に同定されたことが報告されているのみでした。(Giudicessi et al, Hum Mutat 2012, Wang et al, Sezure 2019) 一方、Giudicessiらは、本変異の機能解析を行い、一過性外向きカリウム電流(Ito)密度の増加、不活性化速度の遅延、不活性化からの回復の遅延をきたすことによりIto機能増強、減弱の両方をきたすことが明らかとなりました。 (Giudicessi et al. Hum Mutat 2012) 本変異によるItoの機能増強により心臓チャネル病を、(Olsen et al, Cardiovasc Res 2013, Takayama et al, Heart Rhythm 2019) 機能減弱により脳チャネル病をきたすと考えられ、(Singh et al, Neurobiol Dis 2006, Aronica et al, Neurobiol Dis 2009) 我々は、本変異による心脳チャネル病を新規心脳チャネル病として提唱しました。
てんかん患者は突然死をきたすことが多いとされておりますが、突然死した症例の少なくとも一部は心脳チャネル病による可能性があります。てんかん患者においては心チャネル病を合併している可能性があることから、遺伝子変異を同定し、突然死予防策を講じることにより突然死を未然に防ぐごとが重要と考えられます。
多施設共同研究への参加
AMED研究課題「洞不全症候群の臨床情報・遺伝学的解析に基づくリスク層別化アルゴリズムの開発」(研究開発代表者:国立循環器病研究センター分子生物学 大野聖子先生)の研究開発分担者(中島忠)として参加しております。
過去5年間の論文発表
- 1) Nakajima T, Dharmawan T, Kawabata-Iwakawa R, Tamura S, Hasegawa H, Kobari T, Kaneko Y, Nishiyama M, Kurabayashi M. Biophysical defects of an SCN5A V1667I mutation associated with epinephrine-induced marked QT prolongation. J Cardiovasc Electrophysiol. in press.
- 2) Nakajima T, Kaneko Y, Dharmawan T, Kurabayashi M. Role of the voltage sensor module in Nav domain IV on fast inactivation in sodium channelopathies: The implication of closed-state inactivation. Channels. 2019; 13: 31-343.
- 3) Dharmawan T, Nakajima T, Iizuka T, Tamura S, Matsui H, Kaneko Y, Kurabayashi M. Enhanced closed-state inactivation of mutant cardiac sodium channels (SCN5A N1541D and R1632C) through different mechanisms. J Mol Cell Cardiol 2019;13:88-95.
- 4) Dharmawan T, Nakajima T, Ohno S, Iizuka T, Tamura S, Kaneko Y, Horie M, Kurabayashi M. Identification of a novel exon3 deletion of RYR2 in a family with catecholaminergic polymorphic ventricular tachycardia. Ann Noninvasive Electrocardiol. e12623; 2019.
- 5) Nakajima T, Sugawara T, Kaneko Y, Kurabayashi M. Atrioventricular block-induced torsades de pointes associated with KCNQ1-G269S. Intern Med 2018; 57: 905-906.
- 6) Yoshida K, Nakajima T, Kaneko Y, Kurabayashi M. Implication of Left Bundle Branch Block-Related Cardiac Memory in the Initiation of Torsades de Pointes. J Cardiovasc Electrophysiol. 27: 757-758, 2016
- 7) Nakajima T, Kaneko Y, Saito A, Ota M, Iijima T, Kurabayashi M. Enhanced fast-inactivated state stability of cardiac sodium channels by a novel voltage sensor SCN5A mutation, R1632C, as a cause of atypical Brugada syndrome. Heart Rhythm 12: 2296-2304, 2015
- 8) Nakajima T, Kaneko Y, Kurabayashi M. Unveiling specific triggers and precipitating factors for fatal cardiac events in inherited arrhythmia syndromes. Circ J 79: 1185-1192, 2015


